Geometry Optimization
Determine stable molecular structures by systematically minimizing forces on atoms, enabling prediction of molecular shapes and energetic minima essential for chemical design.
Key Features
.svg)

.jpg)
Elevate Your Research
Find Stable Structures
Locate minimum energy molecular geometries efficiently.
Validate Results
Confirm optimization quality through vibrational analysis.
.webp)
Guide Design
Predict stable molecular arrangements for new compounds.
Increase Efficiency
Automated workflows streamline structure determination.
Technical Specifications
Methodology
Geometry optimization employs gradient-based methods to locate stationary points on molecular potential energy surfaces. The method combines electronic structure calculations with efficient optimization algorithms to minimize atomic forces and energy.
This approach enables reliable determination of equilibrium structures while maintaining the ability to handle various types of constraints when needed.
.svg)
.webp)
Performance Enhancements
Advanced Hessian update schemes accelerate convergence. Pre-optimization workflows using methods such as xTB and HF-3c provide high-quality starting geometries, reducing optimization effort for larger and more complex systems.
Outputs
- Optimized Structure: Final molecular geometry representing the minimum energy arrangement of atoms.
- Convergence Data: Detailed information about optimization progress including energy and gradient evolution.
- Vibrational Analysis: Normal mode frequencies and thermodynamic properties when requested.
- Constrained Optimized Structures: Optimized geometries suitable as starting points for transition state calculations.
.jpg)
Industry Applications
.webp)
Pharmaceuticals
Structure optimization enables prediction of drug molecule conformations and binding geometries. Understanding stable molecular arrangements supports rational drug design and formulation development.
Materials Science
Accurate molecular geometries guide materials development and property prediction. Structural insights enable rational design of materials with targeted characteristics.
.png)
Scalability and User Experience
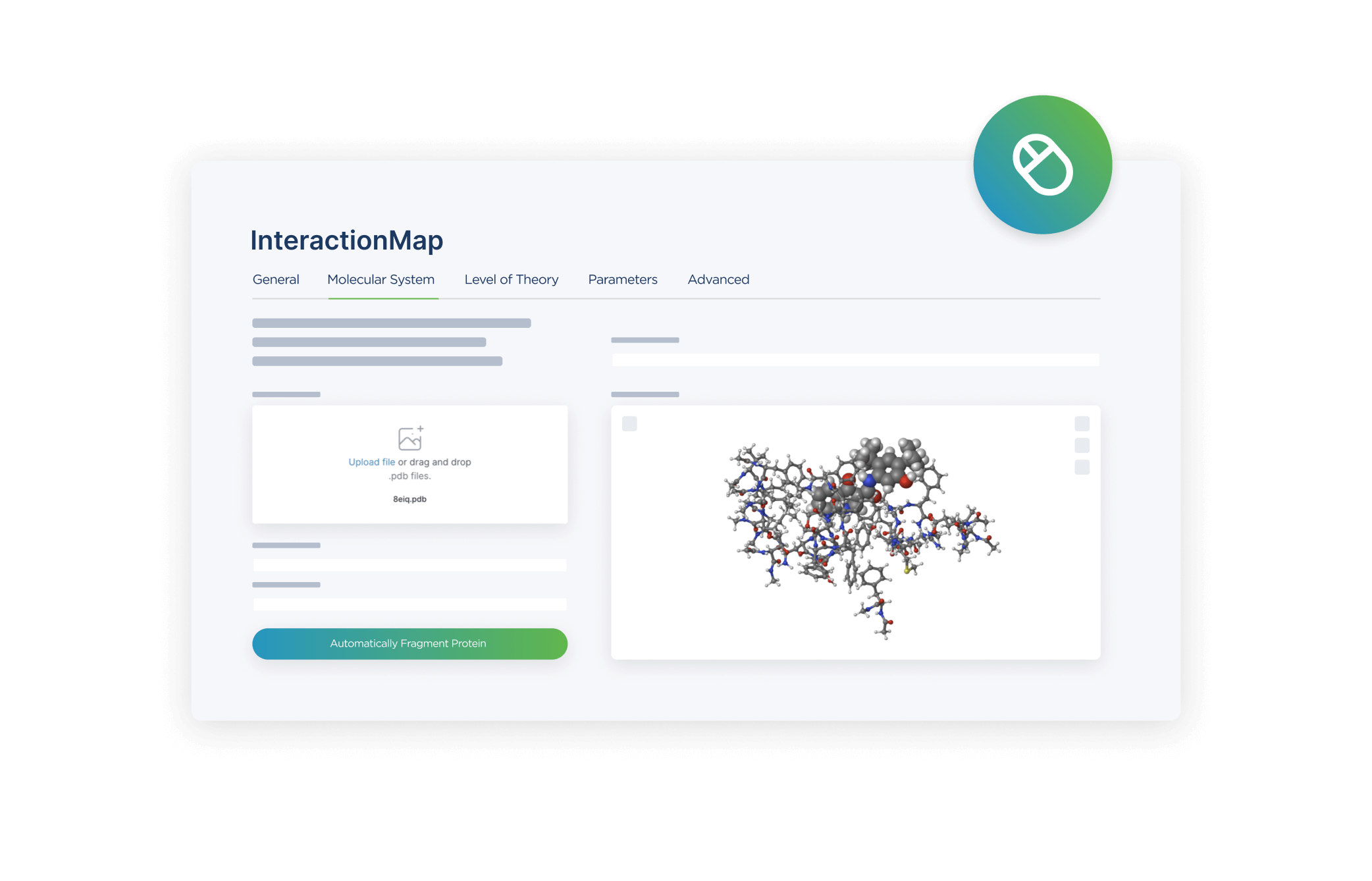
User Interface
An intuitive interface guides optimization setup and analysis. Interactive tools help visualize structural changes and convergence behavior.
.webp)
Scalability
Cloud infrastructure enables efficient processing of multiple structures simultaneously. Batch capabilities support systematic optimization of molecular libraries.
Frequently Asked Questions (FAQs)
Common questions about geometry optimization.

A computational method that determines the most stable arrangement of atoms in a molecule by minimizing its energy.
The method systematically adjusts atomic positions using gradient information to find the lowest energy structure.
Results include optimized molecular structures, energy values, and optional vibrational frequencies and thermodynamic properties.
The method handles various molecular systems from small organic molecules to large biomolecules, with optional constraints available.
Understanding stable structures helps predict molecular properties and behavior in different environments.
Choice of theoretical method, starting geometry quality, and inclusion of environmental effects influence result reliability.